Medical Device Classification and Medical Software Classification
Introduction
Each regulatory agency has defined several different classifications for medical devices. The classifications are, for the most part, related to the perceived risk of the product type. Knowing your medical device classification matters for the following reasons:
1. Product classification will determine what you must do before you can sell your product.
2. Product classification will help you establish requirements during the product development phase, specifically design controls.
3. Product classification is an important component in determining how much it will cost to bring your device to market and give you some idea of how long it will take.
Figure 1 - FDA Device Classification
The medical device classification should not be confused with the software safety class which determines the rigor of software development and level of documentation as defined within IEC 62304 [1] or the “Level of Concern” as defined by the FDA [2]. Both [1] and [2] distinguish three different categories of medical device software and the following sections will expand on these and provide a comparison between them.
EU Requirements for Medical Software Development and Software Safety Classification
IEC 62304 [1] defines the life cycle requirements for medical device software. The set of processes, activities, and tasks described in this standard are recognized by both the EU and US regulatory bodies. It also provides a definition of the software safety classes which the EU uses to establish the documentation required for a submission. Figure 2 below provides an overview of the software development process as defined with IEC 62304 [1].
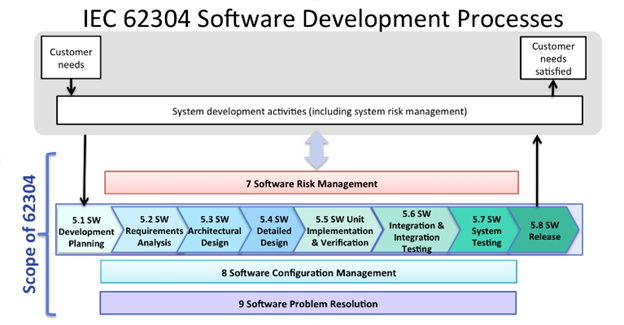
Figure 2 – Overview of software development PROCESSES and ACTIVITIES
Figure 3 below provides a definition of the software safety classes from the IEC 62304 [1]. “The purpose of the software safety classification is to determine the required rigour of the software PROCESSES prior to their start. The software safety class is determined by EVALUATION of the RISK and severity of HARM related to potential SOFTWARE SYSTEM failures and considering specified RISK CONTROLS external to the SOFTWARE SYSTEM” [1].

Figure 3 - IEC 62304 Software Safety Class Definitions
IEC 62304 [1], Figure 3 – Assigning software safety classification is a workflow for establishing the Software Class for a product. The important thing to note is that the determination of safety class is done based upon the RISK of HARM to the patient, operator, or other people. IEC 62304 [1] Table A.1 (reproduced below as Table 1) Summary of requirements by software safety class provides full details of which sections apply for each safety class. Table 2 below shows the documentation required for the different safety classes, for example for class A software no software architecture (chapter 5.3) is required.
NOTE: This approach is different to the requirements of the FDA which is discussed below.

Table 1 - Summary of Requirements by Software Safety Class from IEC 62304 [1].
Table 2 below provides the section headings for the clauses provided in Table 1 above. For any missing subheadings please refer to [1].
Section Name for Clause Clause
| General requirements | 4 |
| Software development planning | 5.1 |
| Software requirements analysis | 5.2 |
| Software ARCHITECTURAL design | 5.3 |
| Software detailed design | 5.4 |
| SOFTWARE UNIT implementation | 5.5 |
| Software integration and integration testing | 5.6 |
| SOFTWARE SYSTEM testing | 5.7 |
| Software release | 5.8 |
| Software maintenance PROCESS | 6 |
| Analysis of software contributing to hazardous situation | 7.1 |
| RISK CONTROL measures | 7.2 |
| VERIFICATION of RISK CONTROL measures | 7.3 |
| RISK MANAGEMENT of software changes | 7.4 |
| Software configuration management PROCESS | 8 |
| Software problem resolution PROCESS | 9 |
Table 2 – Section Name and Number for Clauses IEC 62304 [1].
FDA Requirements for Medical Software Development and Software Level of Concern
As stated previously the IEC 62304 [1] standard is recognized by the FDA for medical software development. The FDA [2] however defines a “Level of Concern” which corresponds to the definitions in IEC 62304 [1] – see Fig 3 below.

Figure 3 - Level of Concern Definitions
The FDA [2] uses the “Level of Concern” to determine the level of documentation required for a submission and does not influence the documentation to be compiled. If you market your products in the US, the software safety class is irrelevant, and you must document according to class C. Do not confuse compilation and submission of documents. FDA [2] contains Table 1 Major Level of Concern and Table 2 Moderate Level of Concern contain several questions which aid in the establishment of the level of concern. These are reproduced below as Tables 3 and 4 within this document. If the answer to any of the questions in Table 3 are yes, then it is a Major Level of Concern otherwise answer questions in Table 4. If any of the answers to the questions in Table 4 is yes, then it is a Moderate Level of Concern. If all the answers to Table 3 and Table 4 is no, then it is a Minor Level of Concern.
| Table 1 Major Level of Concern If the answer to any one question below is Yes, the Level of Concern for the Software Device is likely to be Major. |
| 1. Does the Software Device qualify as Blood Establishment Computer Software? (Blood Establishment Computer Software is defined as software products intended for use in the manufacture of blood and blood components or for the maintenance of data that blood establishment personnel use in making decisions regarding the suitability of donors and the release of blood or blood components for transfusion or further manufacture.) |
| 2. Is the Software Device intended to be used in combination with a drug or biologic? |
| 3. Is the Software Device an accessory to a medical device that has a Major Level of Concern? |
| 4. Prior to mitigation of hazards, could a failure of the Software Device result in death or serious injury, either to a patient or to a user of the device? Examples of this include the following: |
| a. Does the Software Device control a life supporting or life sustaining function? |
| b. Does the Software Device control the delivery of potentially harmful energy that could result in death or serious injury, such as radiation treatment systems, defibrillators, and ablation generators? |
| c. Does the Software Device control the delivery of treatment or therapy such that an error or malfunction could result in death or serious injury? |
| d. Does the Software Device provide diagnostic information that directly drives a decision regarding treatment or therapy, such that if misapplied it could result in serious injury or death? |
| e. Does the Software Device provide vital signs monitoring and alarms for potentially life threatening situations in which medical intervention is necessary? |
Table 3 - FDA Table 1 Major Level of Concern Questions [2]
| Table 2 Moderate Level of Concern If the Software Device is not Major Level of Concern and the answer to any one question below is Yes, the Level of Concern is likely to be Moderate . |
| 1. Is the Software Device an accessory to a medical device that has a Moderate Level of Concern? |
| 2. Prior to mitigation of hazards, could a failure of the Software Device result in Minor Injury, either to a patient or to a user of the device? |
| 3. Could a malfunction of, or a latent design flaw in, the Software Device lead to an erroneous diagnosis or a delay in delivery of appropriate medical care that would likely lead to Minor Injury? |
Table 4 - FDA Table 2 Moderate Level of Concern Questions [2]
Table 5 below shows the submission documentation required for the “Level of Concern” by the FDA.
| Software documentation | Minor | Moderate | Major |
| Level of concern | x | x | x |
| Software description | x | x | x |
| Device hazard analysis | x | x | x |
| Software requirements specification | (x) | x | x |
| Architecture design chart | x | x | |
| Software design specification | x | x | |
| Traceability analysis | x | x | x |
| Description of software environment | x | x | |
| Verification and Validation documentation | (x) | x | x |
| Revision level history | x | x | x |
| Unresolved anomalies | x | x |
Table 5 – Submission Documentation required for the “Level of Concern” by the FDA.
Comparison between EU and FDA
Besides the differences in terms of processes and documentation described above there is the question of the initial “Software Safety Class” for IEC62304 or “Level of Concern” for the FDA. IEC 62304 [1] permits a reduction of the software safety class by means that are external to the software only. Examples are:
- Physical hardware e.g., a stopper.
- Other component containing hardware (electronics) and even software e.g., a watchdog.
- User e.g., responding to a warning (not in IFU) or pressing an emergency stop.
What this means is that by reducing the safety class we can reduce the level of documentation (and the rigor of software development) we need to submit for IEC 62304 [1]. The FDA expects measures to mitigate risks. However, the levels of concern are determined prior to these measures. I.e., a reduction of the level of concern does not impact the documentation to be submitted. The FDA expects you do develop your software as a Major Level of Concern (equivalent to Class C in IEC 62304 [1]) and your submission must include a document for “Level of Concern” which establishes the rest of the submission documentation. The IEC 62304 [1] process is recognised by the FDA and to ease things it is recommended that the process is the one followed. The EU on the other hand expects you do develop your software in accordance with the identified “Safety Class” and your submission must include a document for “Intended Use”. The safety class establishes the rigor for development and the level of documentation required. The IEC 62304 [1] process is recognised by the EU and to ease things it is recommended that the process is the one followed.If it is intended to submit to both the EU and FDA, then the easiest path is to develop software to “Class C” and to produce the “Intended Use” and “Level of Concern” documents and to submit the appropriate documents to the relevant regulatory body.
Introduction to SaMD and its Software Development Process
FDA [3] elaborates upon software for medical and non-medical purposes. “As technology continues to advance all facets of health care, software has become an important part of all products, integrated widely into digital platforms that serve both medical and non-medical purposes. Software, which on its own is a medical device – Software as a Medical Device – is one of three types of software related to medical devices. The other two types of software related to medical devices include software that is integral to a medical device (Software in a medical device) and software used in the manufacture or maintenance of a medical device.” The term Software as a Medical Device is defined by the IMDRF as "software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device." [11] The IEC 82304-1:2016 [5] applies for developing such software and the abstract states: “IEC 82304-1:2016 [5] applies to the safety and security of health software products designed to operate on general computing platforms and intended to be placed on the market without dedicated hardware, and its primary focus is on the requirements for manufacturers. It covers the entire lifecycle including design, development, validation, installation, maintenance, and disposal of health software products.” IEC 82304 [5] utilizes IEC 62304 [1] and effectively builds upon and hence the software is still developed by following IEC 62304 [1] – see Fig 4 below and A.2 Requirements for HEALTH SOFTWARE PRODUCTS in [5].
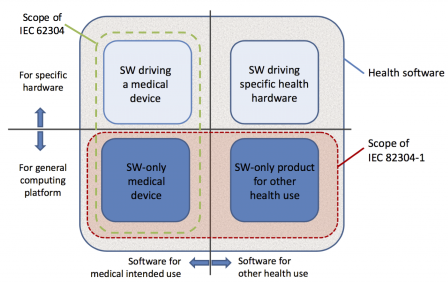
Figure 4 – HEALTH SOFTWARE application domains and scope of related standards [5]
SaMD software is hence developed by following IEC 82304 [5] and IEC 62304 [1] and the same artifacts required previously under IEC 62304 are still required. IEC 82304 has additional requirement to produce artifacts which will have informally been produced by companies in the past.
EU SaMD Device Classification
Figure 5 depicts the rules within the MDR 2017/745 [4] which classify an active device.
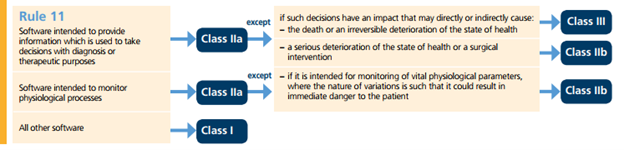
Figure 5 - Active Devices Rules Classifications EU MDR 2017/745
Figure 6 depicts Rule 11 which applies for SaMD device classifications.
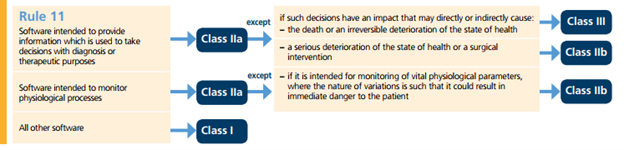
Figure 6 - Graphical presentation of Rule 11 – Active Devices
The SaMD device classification (IIa, IIb or III) defines the conformity assessments and submission requirements according to chapter V in MDR 2017/745 [4].
FDA SaMD Device Classification
SaMD are treated as common medical devices and will follow the premarket requirements as defined by FDA. They are categorized into one of three classes (I, II, or III), based on the degree of risk they present. As device class increases from class I to class II to class III, the regulatory controls also increase, with class I devices subject to the least regulatory control, and class III devices subject to the most stringent regulatory control. The classes of devices, regulatory controls and submission types are summarized in Figure 7.
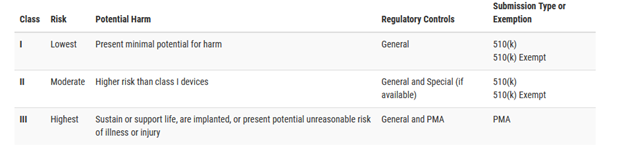
Figure 7- FDA Device Class versus Submission Type
Comparison between EU and FDA for SaMD
In terms of the software development process and independent of the different SaMD device classifications for EU and FDA highlighted above, both IEC 62304 [1] and IEC 82304 [5] apply and the level of documentation that has been described previously for [1] still applies as well as some additional activities and artifacts that are mandated by [5]. Besides developing compliant SaMD Software according to IEC 62304 [1], it may be essential to consider other legislations, standards and guidance documents based on the intended use of the SaMD related to:
- Cybersecurity (FDA Guidance, EU Guidance, AAMI TIR 57 [7])
- Data Privacy (GDPR (EU) , HIPAA (US))
When is an App a Medical Device?
Definition by EU:

There are several useful guidance documents from the EU available as help for defining when an App is a medical device, like the MHRA guidance document “Medical device stand-alone software including apps” [6].
Definition by FDA:
The FDA [9] provides the following definitions for Mobile Apps and Medical Mobile Apps: “Mobile apps are software programs that run on smartphones and other mobile communication devices. They can also be accessories that attach to a smartphone or other mobile communication devices, or a combination of accessories and software. Mobile medical apps are medical devices that are mobile apps, meet the definition of a medical device, and are an accessory to a regulated medical device or transform a mobile platform into a regulated medical device.” Examples of apps that are classified as medical devices:
- Apps that are an accessory or an extension of a medical device. Examples are apps which directly control (for example, starting a blood pressure measurement) or apps that remotely display values of medical devices.
- Apps that perform support for individual patients such as drug dose calculations or detection of drug interactions, or contraindications.
- Software for analysis or processing of radiological images (with a diagnostic purpose).
Examples of Mobile Apps That Are NOT Medical Devices [10]:
- Apps complying with an electronic copy of a book or reference work.
- Apps that are used as a training example by clinical staff or patients.
- Apps that serve the payroll.
- Apps that have not been specifically designed for the medical field. For example, a digital magnifier.
Definition by IMDRF:
To classify Apps as medical devices, the IDMRF guidance document [11] suggests four categories (I, II, III, and IV) which are based on the levels of impact on the patient or public health where accurate information provided by the Software as a Medical Device to treat or diagnose, drive, or inform clinical management is vital to avoid death, long-term disability, or other serious deterioration of health, mitigating public health. The Level IV category is Software as a Medical Device with the highest impact on the patient or public health and Level I is the lowest.
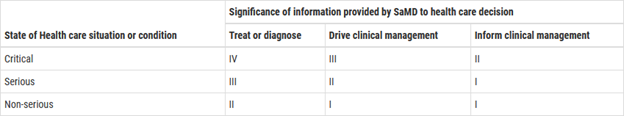
Figure 8 - FDA/IMDRF SaMD Classification
The IMDRF categorization framework is not a regulatory classification, nor implies a convergence of classifications rules. However, it does set a path towards common vocabulary and approach. Additional work is required to align existing classification rules with this framework. The categorization framework is not meant to replace or conflict with the content and/or development of technical or process standards related to software risk management activities.
References
[1] IEC 62304:2006/Amd 1:2015 Medical device software — Software life cycle processes — Amendment 1
[2] Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices -https://www.fda.gov/media/73065/download
[3] https://www.fda.gov/medical-devices/digital-health-center-excellence/software-medical-device-samd
[4] MDR 2017/745 - REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC
[5] IEC 82304-1:2016 Health software — Part 1: General requirements for product safety
[6] MHRA guidance document “Medical device stand-alone software including apps” https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/957090/Software_flow_chart_Ed_1-07b-UKCA__002__FINAL.pdf
[7] MDCG 2019-11 Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR October2019
[8] AAMI TIR57:2016 Principles for medical device security—Risk management
[9] Device Software Functions Including Mobile Medical Applications https://www.fda.gov/medical-devices/digital-health-center-excellence/device-software-functions-including-mobile-medical-applications
[10] Examples of Mobile Apps That Are NOT Medical Devices https://www.fda.gov/medical-devices/device-software-functions-including-mobile-medical-applications/examples-mobile-apps-are-not-medical-devices
[11] IMDRF - “Software as a Medical Device": Possible Framework for Risk Categorization and Corresponding Considerations http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-140918-samd-framework-risk-categorization-141013.pdf